
Wczoraj szachy, dziś białka, jutro…?
Kolejne sukcesy sztucznej inteligencji. Najpierw komputery zaczęły regularnie ogrywać arcymistrzów w szachach, a następnie nauczyły się grać w go. Teraz wreszcie, po wielu latach prób, algorytm komputerowy nie tylko wygrał konkurs na najdokładniejsze teoretyczne zwijanie białek (CASP), ale wręcz wyprodukował wyniki porównywalne z wynikami eksperymentalnymi. Ba, w “dogrywce” dał radę przewidywać strukturę białka, dla którego wyniku eksperymentalnego jeszcze nie było. Jego przewidywanie było tak trafne, że dzięki niemu z surowych danych (nad którymi naukowcy wcześniej głowili się przez 10 lat) strukturę eksperymentalną uzyskano w pół godziny. Ale o co dokładnie chodzi i dlaczego to jest ważne? Pozwól, że wyjaśnię.
Do czego służą białka?
Białka to cząsteczki składające się z aminokwasów połączonych w szereg – jeden za drugim. Pełnią w organizmach wiele istotnych funkcji. Od prostego budulca, zapewniającego właściwy kształt różnych struktur, przez przyspieszanie reakcji chemicznych (o tym już niedługo na naszym blogu!), aż po ruch czy komunikację pomiędzy komórkami. Żeby je pełnić skutecznie, potrzebują mieć konkretną strukturę trójwymiarową. Luźno merdająca nitka nie będzie przecież skutecznie transportowała jonów przez nieprzepuszczalną błonę. Jeśli z tej nitki utkamy kawałek rury i nim tę błonę przeszyjemy na wylot, to już coś da radę przez nią przeniknąć.
Żeby dowiedzieć się, jak dokładnie białko pracuje (albo naprawić to, które pracuje źle), musimy znać jego strukturę przestrzenną. Innymi słowy – dopiero znajomość trójwymiarowej struktury białek daje nam możliwość projektowania leków, które będą na te białka działać. Ale jak poznać tę trójwymiarową strukturę białek?
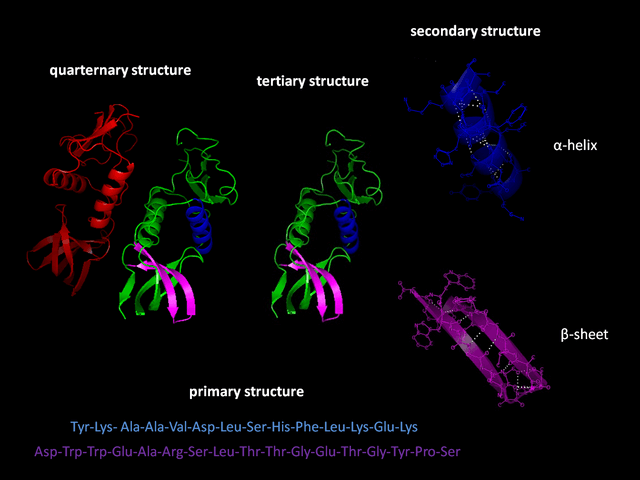
Poznawanie struktury białka
Można przeprowadzić eksperyment. Jest kilka metod, które potrafią przeanalizować białko i powiedzieć nam, jaki jest jego kształt. Są to metody bazujące na krystalografii, mikroskopii elektronowej czy rezonansie magnetycznym1Po chemicznemu nazywa się to NMR – jądrowy rezonans magnetyczny. Po medycznemu, wykorzystane do obrazowania – MRI, obrazowanie rezonansem magnetycznym – głównie po to, żeby nie straszyć pacjentów słowem “jądrowy”. NMR nie ma nic wspólnego z promieniotwórczością, nazywa się tak, jak się nazywa, bo bada jądra atomów.. Te metody wymagają posiadania dość dużych ilości białek, często o wysokiej czystości. Często pojawiają się dodatkowe ograniczenia. Na przykład w metodach bazujących na krystalografii z białek trzeba wyhodować kryształy. Nie zawsze jest to takie proste. Nawet jeśli uda się spełnić wszystkie warunki, analiza danych potrafi być czasochłonna i problematyczna.
Dlatego też od lat starano się w teorii przewidzieć strukturę trójwymiarowych białek na podstawie sekwencji ich kolejnych bloków budulcowych. Co ciekawe, to kolejna dziedzina, w której przez długi czas intuicja człowieka biła na głowę komputery – zapoznajcie się na przykład z grą FoldIt2Środek ciężkości ostatnio przesunął się na inne dziedziny, ale projekt nadal jest rozwijany. Komputery z dużymi białkami nie radziły sobie wcale. Do niedawna najwyższe wyniki osiągane przez algorytmy w stustopniowej skali podobieństwa przewidywań do rzeczywistości oscylowały w okolicach 60 (90 uznaje się za wynik wystarczająco zbliżony do faktycznej struktury).
Sukces DeepMind
I oto w tym roku algorytm AlphaFold, opracowany przez grupę DeepMind, osiągnął średni wynik 92.4. Poprawa od poprzedniej edycji konkursu była tak wielka, że w pierwszej chwili organizatorzy posądzili DeepMind o oszustwo. Algorytm dostał więc dodatkowe zadanie. Miał przewidzieć sekwencję białka, którego strukturę próbowano określić z danych eksperymentalnych przez 10 lat. I z tym zadaniem, jak pisałem we wstępie, AlphaFold poradził sobie śpiewająco. A zdaniem autorów wciąż jest jeszcze miejsce na poprawę. Trzymajmy kciuki, bo informacja o tym, jak wygląda struktura białek, może przyczynić się do dużo lepszego zrozumienia mechanizmów działania życia. A co za tym idzie, do szybkiego rozwoju medycyny i innych pokrewnych nauk.
Żródła:
https://science.sciencemag.org/content/370/6521/1144
https://moalquraishi.wordpress.com/2018/12/09/alphafold-casp13-what-just-happened/
Zainteresowało Cię to, co czytasz? Chcesz wiedzieć więcej? Śledź nas na Facebooku, i – pozwól, że wyjaśnię!
Konsekwentnie nazywasz AlphaFold algorytmem, wyjaśnisz? (you see what I did here? 😉) DeepMind używa wprost „AI system”. A jesli widzisz różnicę między tymi dwoma kategoriami, to już taki np. AlphaStar chyba trudno byłoby nazwać „tylko” algorytmem? Interesuje mnie, co o tym sądzisz.
Hasło „algorytm” jest tu rzeczywiście pewnym uproszczeniem – ale nie jest niepoprawne. System sztucznej inteligencji jest zbiorem algorytmów. Algorytmy te mogą się do pewnego stopnia same modyfikować, żeby dopasować się do zadań stojących przed nimi. Jeszcze inna kwestia to pytanie, w jakim stopniu AlphaFold jest systemem sztucznej inteligencji, a w jakim systemem uczenia maszynowego – on dostaje dane ustrukturyzowane, co sugeruje, że jednak jest to raczej uczenie maszynowe, ale każda pliszka swój ogonek chwali, a hasło „sztuczna inteligencja” brzmi jednak bardziej dostojnie.
[…] jak wspomniałem, również synteza białek na podstawie DNA nie jest taka prosta. Przede wszystkim: białka nie powstają bezpośrednio na […]
[…] białka. W związku z tym, zamiast podawać nam całe patogeny, w szczepionce podaje się ich białka powierzchniowe. Nie mają one szansy na wywołanie choroby, a wystarczają, żeby nauczyć […]
[…] kształt. O tym, że struktura białka definiuje jego właściwości, mogliście już u nas kiedyś przeczytać. Większość białek, jeśli przybierze niewłaściwy kształt, po prostu nie działa. Niektóre […]
[…] się, że może mieć bardzo duży. Wałkowana na lekcjach biologii anemia sierpowata jest właśnie efektem mutacji, która zamienia […]
[…] twórca materiały czerpał z innych źródeł. Interesujące jest to, że szukali w tych próbkach białek. Dlaczego? Ponieważ różne organizmy produkują różne białka, więc dokładna identyfikacja […]